INTRODUCTION
Cleaning validation should be performed to confirm the efficiency of any cleaning procedure when pharmaceutical products are in contact with equipment. In the pharmaceutical manufacturing industry it is well known that the manufacturing equipment and manufacturing area should be cleaned after every manufacturing process of drug products and this process is strictly endorsed by the regulatory authorities. Cleaning validation is a vital analytical responsibility of the quality management system in the pharmaceutical industry and this process ensures that the cleaning procedure effectively eliminates the residue from the manufacturing equipment and manufacturing area below a predetermined tolerable limit. The cleaning process ensures the product quality of different products, is a helpful tool to avoid cross-contamination, and is a requirement of European Union guidelines for Good Manufacturing Practice and the United States Food and Drug Administration (USFDA). Cleaning validation involves two different activities: one is development and validation of the cleaning process used to remove the drug from the manufacturing equipment surfaces and the other is development and validation of the methods used to measure the residues on the surfaces of the manufacturing equipment. Evaluation of the sensitivity and specificity of the analytical method used to detect residue is critical. The residue analytical method should able to detect and quantify the drug substance at a very low level from the manufacturing equipment. The residue analytical procedure should be tested in the mixture of sampling method used to show that residue can be recovered from the equipment surface with the specified levels in the accuracy study before concluding the sampling procedure. In general, two types of sampling procedure were found acceptable by the regulatory authorities and frequently practicing pharmaceutical industries. The popular sampling method is the direct method of sampling on the surface of the manufacturing equipment and another method is to use rinse solutions from the manufacturing equipment. The positive aspect of direct sampling of the equipment surface is that the areas hardest to clean and that are reasonably available can be projected, important for finding a level of residue per given surface area. In the case of rinse samples, the two benefits of using rinse samples are that a larger surface area may be sampled and unreachable systems or ones that cannot be routinely disassembled can be sampled and estimated. The disadvantage of rinse samples is that the residue may not be soluble or may be physically occluded in the manufacturing equipment surface area.
With direct surface sampling there is a possibility of interference from the swab sticks as they have some glue content and before finalizing the sampling procedure the specificity also should be evaluated. The selection of the extraction solvent is a critical step during the development of the cleaning method, the drug substance should be soluble and recoverable across the accuracy swab sample level, and the results should meet the acceptance criteria.
The drug product manufacturer’s rationale for the residue limits established should be logical based on the manufacturer’s scientific knowledge of the materials involved. It is important to describe the analytical method sensitivity of the residue method in order to fix sensible acceptable limits. According to the USFDA, the limit should be based on logical criteria, involving the risk associated with residues of determined products. The calculation of an acceptable limit of residues and a maximum allowable carryover for an active pharmaceutical ingredient in the production equipment should be based on therapeutic doses, toxicity, and a general limit (10 µg). Several mathematical formulas have been proposed to establish the acceptable residual limit.1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17
The drug substance dipyridamole (Figure 1): chemical name: 2,2’,2”,2””-[[4,8-di (piperidin-1-yl) pyrimido [5,4-d] pyrimidine-2,6-diyl] dinitrilo] tetraethanol, CAS Registry number: 58-32-2, molecular formula: C24H40N8O4, molecular mass: 504.6, appearance: bright yellow, crystalline powder, solubility: practically insoluble in water, slightly soluble in acetone, soluble in anhydrous ethanol, and it dissolves in dilute mineral acids.
The aim of the present study was to develop a simple and fast analytical method for the estimation of the dipyridamole content in swab samples after the manufacture of dipyridamole modified release capsules on the surface of the manufacturing equipment and to meet the regulatory requirements. Hence, the developed method was subjected to analytical validation with respect to specificity, linearity, precision, accuracy, robustness, and ruggedness. The specificity studies were performed on the diluent, swab, and placebo during the analytical method validation as per International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human (ICH) guidelines.18 The developed and validated method can be used for the routine swab samples analysis.
MATERIALS AND METHODS
Chemicals and reagents
High performance liquid chromatographic (HPLC)/analytical grade water, potassium dihydrogen phosphate, methanol, and sodium hydroxide were used. The dipyridamole drug substance and dipyridamole working standard were supplied by Bluefish Pharmaceuticals Private Limited (India).
Equipment
The analytical method was developed and validated by using the HPLC from Agilent 1200 with a VWD/PDA detector. The output signal was monitored and processed using specific software. An analytical balance from Mettler Toledo, a Sartorius pH meter, and a refrigerator were used.
Chromatographic conditions
The proposed method was developed using a Hypersil BDS C18 (150×4.6 mm) 5 µm column with mobile phase containing a mixture of mobile phase (buffer: potassium dihydrogen phosphate buffer, pH 7.0±0.05) and methanol solution in the ratio of 30:70 v/v. The flow rate was 1.5 mL/min with a column temperature of 45°C, detection wavelength of 295 nm, and sample injection volume of 5 µL.
Preparation of solutions
Diluent solution
Methanol was used as diluent.
Preparation of dipyridamole standard solution
Weigh and transfer about 50 mg of dipyridamole working standard into a 50 mL volumetric flask. Add about 35 mL of diluent and sonicate for 2 to 3 min until the material is completely dissolved. Pipette out 1 mL of the above solution into a 100 mL volumetric flask, make it up to volume with diluent, and mix well. Pipette out 4 mL of the above solution into a 10 mL volumetric flask, make it up to volume with diluent, and mix well.
Preparation of test tubes and swabs
Take the clean and dry test tubes. Rinse the required number of swabs and test tubes with about 10 mL of swabbing solvent two times. Squeeze out the swab against the side of the test tubes and discard the swabbing solvent.
Preparation of blank solution
Transfer 10 mL of swabbing solvent to the above cleaned test tube. Place a cleaned swab into the test tube and sonicate for 10 min. Squeeze the swab and take it out and mix well.
Preparation of test solution
Transfer 10 mL of swabbing solvent to the above cleaned test tube. Place a cleaned swab into the test tube to wet the swab with swabbing solvent. Squeeze the swab by pressing it against wall of the test tube. Do the swabbing at the prescribed area of equipment. After swabbing, place the swab in the above test tube containing swabbing solvent and sonicate for 10 min.
Squeeze the swab by pressing it against the wall of the test tube and take it out and filter it through a membrane filter and inject.
System suitability criteria
The present relative standard deviation of the dipyridamole peak area for six replicate injections should not be more than 5.0.
The tailing factor for dipyridamole peak in standard solution should not be more than 2.0.
The present relative standard deviation of dipyridamole peak retention time for six replicate injections should not be more than 1.0.
The % recovery for dipyridamole check standard solution should not be less than 95.0% and 105.0%.
RESULTS AND DISCUSSION
Method development
During the method development stage, the standard solution was prepared with a known concentration, blank solution was scanned in a ultraviolet spectrophotometer, and the diluent blank (Figure 2) and dipyridamole working standard (Figure 3) spectrums collected to check the wavelength maxima.
The dipyridamole peak retention time was about 2.8 min in the chromatogram and the relative standard deviation for the six replicate injections was 0.2% and this proved that the method is reproducible. The accuracy study was conducted by spiking a known concentration of dipyridamole solution in an SS plate of 0.4 µg/mL, 4 µg/mL, and 6 µg/mL and the % recovery for all the levels was calculated and the results were found in the range of 99% to 100% and this proved that the method is accurate. The linearity study was conducted by spiking a known concentration of dipyridamole solution of about 0.4 µg/mL, 1 µg/mL, 2 µg/mL, 4 µg/mL, 5 µg/mL, and 6 µg/mL, the square of correlation coefficient was calculated and found to be 0.999, and this proved that the method is linear from 0.4 µg/mL to 6 µg/mL. Based on the above-mentioned satisfactory results, the below-mentioned chromatographic conditions were finalized for the quantitative estimation of dipyridamole in the swab samples from drug product manufacturing of the dipyridamole modified release capsules equipment surface after manufacturing. The chromatographic conditions are a Hypersil BDS C18 (150×4.6 mm) 5 µm column with mobile phase containing a mixture of mobile phase (buffer: potassium dihydrogen phosphate buffer, pH 7.0±0.05) and methanol in the ratio of 30:70 v/v. The flow rate was 1.5 mL/min with column temperature of 45°C and detection wavelength of 295 nm. The injection volume was 5 µL with isocratic flow. Hence, this method can be introduced for routine swab analysis.
Method validation
The proposed analytical method for the quantitative estimation of dipyridamole in swab samples from drug product manufacturing of the dipyridamole modified release capsules equipment surface area after manufacturing was validated as per the ICH.18 The validation characteristics specificity, precision, accuracy, linearity, range, ruggedness, and robustness were determined.
System suitability
To check the system suitability criteria, solutions were prepared and injected as per the test method of analysis. All the system suitability parameters were found well within the acceptance criteria. A summary of the system suitability is given in Table 1.
Specificity
To study the specificity, required solutions like diluent as blank, swab blank, and standard solution were prepared and injected as per the test method. It was observed that there was no peak interference at the retention time of dipyridamole from blank and swab blank solutions in the chromatogram. Specimen chromatograms of diluent as blank are shown in Figure 4, swab blank in Figure 5, and standard in Figure 6, and overlaid chromatograms of diluent as blank, swab blank, and standard in Figure 7.
Estimation of limit of detection (LOD) and limit of quantification (LOQ)
To evaluate the concentration limits as LOD and LOQ, a series of linearity solutions were prepared ranging in concentration from about 0.1 µg/mL to 2.0 µg/mL and the square of correlation coefficient, slope of the curve, and y-intercept were determined. The LOQ was calculated based on the standard deviation of the response and the slope as mentioned in the formula below. A summary of the results of the LOD and LOQ estimation study is given in Table 2 and Figure 8.
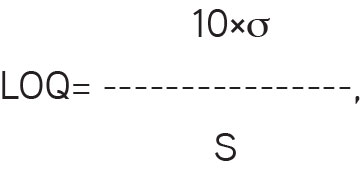
where
σ=the standard deviation of the response
S=the slope of the calibration curve
Method precision at LOQ level
The method precision at LOQ concentration was determined by preparing six replicate test preparations (n=6) of dipyridamole stock solution and samples were analyzed as per the test method. The % relative standard deviation (RSD) for response of dipyridamole six replicate injections was calculated and found within the acceptance criteria. A summary of the method precision for the LOQ level study results is given in Figure 9.
Method precision (repeatability)
Precision was determined by preparing six replicate test preparations (n=6) of dipyridamole stock solution spiked onto an SS plate (4×4 inch) and samples were analyzed as per the test method. The % recovery for replicate injections and % RSD for response of six replicate injections of dipyridamole were calculated and found within the acceptance criteria. A summary of the method precision study results is given in Figure 10.
Accuracy
In the accuracy study, a series of sample solutions were prepared in triplicate six preparations for lower level (LOQ) and higher level (500%) by spiking the dipyridamole drug substance stock onto an SS plate (4×4 inch) at LOQ 50%, 100%, 200%, 300%, and 500% and analyzed as per the test method. The spiked concentrations of dipyridamole were 0.12 µg/mL, 2.01 µg/mL, 4.03 µg/mL, 8.05 µg/mL, 12.08 µg/mL, and 21.14 µg/mL. Individual % recovery, mean % recovery, % RSD, and squared correlation coefficient for linearity of the test method were calculated and the results were found within the predefined acceptance criteria. A summary of the accuracy study results is given in Table 3 and Figure 11.
Linearity
The linearity was studied by analyzing the standard solutions. A series of solutions of dipyridamole standard solutions were prepared in the range of LOQ to about 500% and injected into the HPLC system. Linearity of detector response was established by plotting a graph of concentration vs. response of dipyridamole. The detector response was found to be linear from about LOQ to 500% and injected into the HPLC system and analyzed as per the test method. The concentrations of dipyridamole were 0.1208 µg/mL, 2.0137 µg/mL, 4.0274 µg/mL, 8.0548 µg/mL, 12.0821 µg/mL, and 20.1369 µg/mL.
The square of correlation coefficient, slope, and % y-intercept at 100% level, and intercept and residual sum of squares were calculated and the results met the acceptance criteria. A summary of the linearity study results is given in Table 4 and Figure 12.
Ruggedness
The intermediate precision was determined by preparing six replicate test preparations (n=6) of dipyridamole stock solution spiked onto an SS plate (4×4 inch) and samples were analyzed as per the test method using a different HPLC system and a different column of the same make by a different analyst on a different day. The % recovery for replicate injections and % RSD for response of six replicate dipyridamole injections were calculated and met the acceptance criteria. A summary of the ruggedness study results is given in Table 5 and Figure 13.
Solution stability and mobile phase stability
The solution stability of dipyridamole was determined by keeping swab sample solution and standard solutions in tightly capped volumetric flasks at room temperature for 1 day and 2 days and measuring against freshly prepared standard solution. The standard solution and swab sample solutions was found stable for 2 days at room temperature.
The stability of the mobile phase was also determined by freshly prepared solutions of dipyridamole at 1 day and 2 days. The mobile phase was found stable for 2 days at room temperature.
Robustness
Robustness of the proposed method was performed by keeping the chromatographic conditions constant with the following deliberate variations:
i. Change in flow rate
ii. Change in wavelength
iii. Change in mobile phase buffer pH
iv. Change in HPLC column temperature
v. Change in mobile phase composition
The standard solution was injected in replicate for each above mentioned change. The system suitability parameters were recorded for dipyridamole peak and the system suitability found well within the acceptance criteria. A summary of the robustness study results is given in Table 6.
The authors declare that no experiments were conducted using human subjects and no ethics committee approval is required for this publication.
APPLICATION OF THE PROPOSED METHOD
The developed analytical method can be applied for the analysis of swab samples from the dipyridamole drug product manufacturing unit. All the analytical validation parameters met the predefined acceptance criteria and the method was proven to be suitable for analysis of swab samples from drug product manufacturing of the dipyridamole modified release capsules equipment surface area after manufacturing.
CONCLUSIONS
The developed method was validated as per the ICH guidelines and can be used for the quantitative estimation of dipyridamole in swab samples from drug product manufacturing of the dipyridamole modified release capsules equipment surface area after manufacturing. The method was precise, accurate, linear, robust, rugged, and specific and there was no interference found during the specificity study at the retention time of dipyridamole peak. The validated method can be applied for swab samples from drug product manufacturing of the dipyridamole modified release capsules equipment surface area after manufacturing.
ACKNOWLEDGMENTS
The authors wish to thank the management of Bluefish Pharmaceuticals Private Limited for their support and encouragement.
Conflicts of interest: No conflict of interest was declared by the authors. The authors alone are responsible for the content and writing of the paper.